Volv, supported by Sanofi, and working with Optimum Patient Care, and collaborating with a specialist Consultant Clinician, is performing research to build algorithms in the UK which are aimed at finding ways to better identify people living with Fabry or Pompe disease.
This novel and innovative methodology, inTrigue, is highlighting ways in which we can be much more precise in detecting people living with either disease much earlier.
Are you a Fabry or Pompe specialist in the UK and want to know more, or collaborate? Please contact us.

inTrigue: helping people living with disease get better outcomes
In the sections below you will find an overview of how we create models to help predict which people might be at risk of disease, some of the current performance metrics, and also some background information on both Fabry and Pompe disease.
By using the inTrigue methodology in collaboration with Optimum Patient Care (OPC) in the UK and the OPC Research Database and supported by Sanofi, we are learning novel patterns of disease, we do this because using published medical criteria does not help find the patients that remain undiagnosed and in fact highlights many more patients that do not in fact have disease (false positives).
The inTrigue approach looks for people that cannot be found using those methods. inTrigue is designed to help clinicians detect the people who are living with a rare or difficult-to-diagnose disease and help uncover those people who are therefore otherwise unlikely to get a diagnosis.
Importantly, this is a research project that
- focusses on a limited population at first
- works with a population of clinicians that have signed up for the OPC quality improvement (QI) programme to improve the quality of care for patients in general practice
- aims to use the feedback from clinicians to improve the approach
This is a completely different level of performance that promises to reduce the time to a diagnosis, and also importantly, uncover the undiagnosed patients.
OPC quality improvement (QI) programme:
(https://www.primescholars.com/articles/strategies-that-promote-sustainability-in-quality-improvement-activities-for-chronic-disease-management-in-healthcare-se-100520.html)

Volv, Sanofi and OPC: collaborating for people living with disease
Volv, supported by Sanofi, and leveraging the data from OPC in the UK, is creating a unique collaboration that does not stop here.
Introduction
The first phase of this project was to collaborate to build new types of models for two rare diseases: Fabry and Pompe. To do this, we focussed on primary health care records, i.e. the records that general practitioners use.
Both diseases are difficult to diagnose for primary care clinicians, and as a result, remain underdiagnosed. For Pompe disease in the UK, it is estimated that 50% of people with the disease are not being diagnosed, leading to a longer delay until they eventually do get diagnosed. This data is managed by Optimum Patient Care, which provides de-identified data, of around 8.5 million patient records, for research purposes. Data security and protection are paramount. This means that the data remains anonymous and secure during the disease model development process.
The data complies with:
- GDPR/ DPA 2018 compliant
- Secured EHR data extraction
- Data is de-identified (no PID)
- Data is pseudonymised SHA256
- Secure data encryption AES256
- Secure data transfer via HSCN
- NHS DSP Toolkit (ref: 8HR5)
- Non-identifiable data is contributed to OPCCRD for ethically approved research
- NHS IHRA REC (ref: 20/EM/0148)
Phase 1: Learn an algorithm/model for the diseases and validate with expert clinicians
The first phase of the inTrigue methodology involved an iterative process of finding a way to determine what makes patients with Fabry and Pompe disease stand out from all other patients. We used a combination of data science (or AI) approaches to get to a list of patients that plausibly have a disease.
Within this phase, crucially and differentiatingly, we also needed to validate whether the approach has worked by checking the inTrigue results with an expert clinician. We did this with a consultant in a specialist Fabry and Pompe department in a UK teaching hospital. The results of this evaluation can be seen in the results section.
Once the clinician’s validation was complete, we then take those inputs and optimise the algorithm, which will again boost the performance. Once this is done, we are ready to move to Phase 2.
Phase 2: Clinical follow-up on plausible patients, more accurately and earlier
In this second phase, the algorithm is applied to the data, and clinicians are asked if they want to participate in the model deployment programme. The clinicians need to give their consent to be part of this quality improvement programme. Several QI programmes are already in place and if they agree, they can then check to see if any of the patients in their practice are at risk of these diseases. This is done through the remote installation of reports in the GP system. We can then monitor to see if there is an improvement in terms of quality of clinical care.
More results on this aspect of the deployment of the models will be published at a later stage, but the optimisation steps post clinician validation shows significant improvement on these results presented here.
Later phases
After this programme, consideration is being given to deploying the models more widely by embedding them into GP systems nationwide.
Initial metrics on model performance
Model performance: Fabry disease in UK
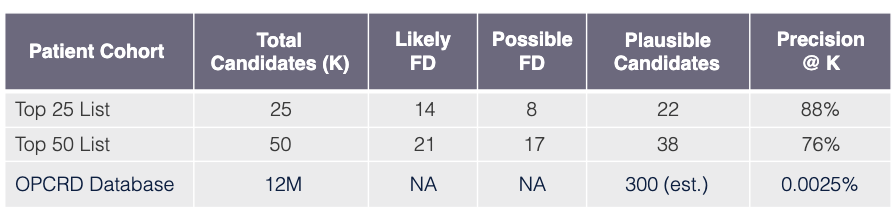
Task
Use model learned via Algorithm SLSL to find undiagnosed FD patients in OPCRD EHR database GP-EHR-DB-UK (18M patients).
Evaluation procedure
Request that FD specialist practicing in UK review EHRs of top 50 candidate patients (candidates have predicted probabilities exceeding FD threshold FD).
Evaluation outcome
Results are very promising showing that out of 50 patients the top 25 have a precision of 88%, and when the total 50 patients are considered the precision remains high at 76% using the precision@k metric.
Model performance: Pompe disease in UK
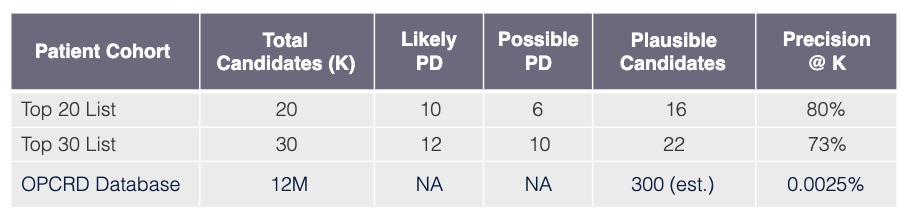
Task
Use model learned via Algorithm SLSL to find undiagnosed PD patients in OPCRD EHR database GP-EHR-DB-UK (18M patients).
Evaluation procedure
Request that PD specialist practicing in UK review EHRs of top 30 candidate patients (candidates have predicted probabilities exceeding PD threshold xPD).
Evaluation outcome
The results for Pompe are also very promising showing that out of 30 patients the top 20 have a precision of 80%, and when the total 30 patients are considered the precision remains high at 73% using the precision@k metric.
Refinement of models post clinical validation step
In the refinement steps following the clinical review phase, we see a significant performance boost. However, these results tend to have an upward bias, so we do not report them here. Instead, these results will be tested out as we deploy the models in this next phase of the programme.
What is Fabry disease?
Fabry disease is closely related to mucopolysaccharidoses and is one of the lysosomal storage diseases. It was first described in 1898 by William Anderson and Johannes Fabry and is also referred to by some as Anderson–Fabry disease.
What causes Fabry?
In the course of normal life there is a continuous recycling process in the body which consists of building new materials and breaking down old ones ready for disposal. This activity takes place in a special part of the body’s cells called the lysosome. This process requires a series of biochemical tools called enzymes. The enzyme alpha-galactosidase A (alpha-GAL) is essential in breaking down the fatty acid globotriaosylceramide (GL3).
People with Fabry cannot make enough of alpha-GAL, without enough levels of the enzyme the normal functioning of vital organs is affected. When GL3 is not completely broken down it builds up within the cells of the body causing progressive damage. Organs such as kidney, heart and brain eventually start to deteriorate, and severe or life-threatening complications can arise. Babies may show little sign of the disease but as more and more cells become damaged by an accumulation of these waste products, symptoms start to appear.
A long road to diagnosis
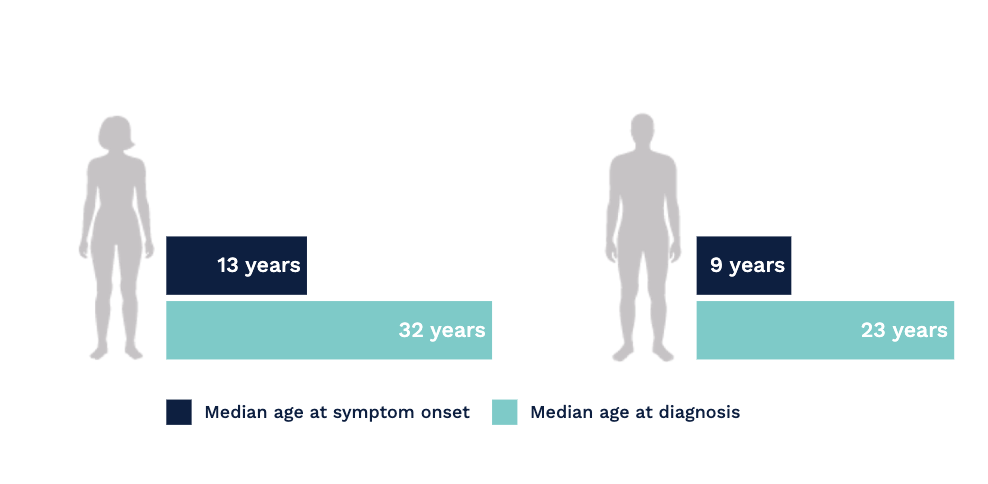
In a study based upon the Fabry Registry, due to the nonspecific and heterogeneous nature of early symptoms in Fabry disease, diagnostic delays and misdiagnosis were common.[1]
Fabry disease is often misdiagnosed and confused with rheumatoid or juvenile arthritis, rheumatic fever, erythromelalgia, Raynaud’s syndrome, neurosis, lupus, acute appendicitis, or multiple sclerosis. Pain, particularly in children, can be dismissed as malingering.[2]
Connecting seemingly unrelated symptoms to Fabry disease can help avoid diagnostic delays and help patients receive disease management sooner.[1]
Importance of Screening for Fabry Disease in High-Risk Populations.
Although Fabry is considered a rare disease, the prevalence of Fabry disease in patients with certain disorders, such as unexplained chronic kidney disease and hypertrophic cardiomyopathy, may be higher than in the general population.[3,4] Consequently, it is important to screen patients with these conditions for Fabry disease.
References
- Eng CM, et al. J Inherit Metab Dis. 2007;30(2):184–192.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1–49.
- Terryn W et al. Nephrol Dial Transplant. 2013; 28:505–517.
- Elliot PM, et al. Eur Heart J. 2014; 35 (39): 2733–2779.
What is Pompe disease?
Pompe disease is a disorder of the metabolism first described in 1932 by Dr J C Pompe. In this case it means that there is a problem with one of the stages in the normal processing of food to make energy.
It is a rare neuromuscular, genetic condition that occurs in babies, children and adults who inherit a defective gene from each of their parents. Though the genetic defect that causes GSD2 is present at birth, symptoms may appear at any time from birth to adulthood.
Pompe disease is an ultra-orphan disease that is currently diagnosed in less than 200 people in the United Kingdom.
Pompe disease (glycogen storage type II disease) is caused by a deficiency of acid alphaglucosidase (GAA) enzyme activity, resulting in lysosomal glycogen accumulation in muscles and irreversible muscle damage.[1-3]
Hidden, undiagnosed populations

Epidemiology
Incidence estimates for Pompe disease range from 1 in 33,333 to 1 in 138,000 [1-2]. However, it is difficult to know exactly how many people are actually affected.
It is estimated that the current worldwide prevalence may be 1 in 57000.[3]
However, it is thought that around 50% of the patients in the UK remain undiagnosed [4]
References
- Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan Screening Program. Pediatrics 2008; 122: e39–45.
- Ausems MGEM, Verbiest J, Hermans MMP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in the Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999; 7: 713–716.
- AGSD, Pompe Disease (GSD2). Website, available online here: https://agsd.org.uk/all-about-gsd/gsd-variants/pompe-disease-gsd2 / [Last accessed October 2019]
- Ausems MGEM, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJJ, Wokke JHJ. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 1999; 52: 851–853.





